
Our research is focused on understanding the molecular basis of quiescence using in vitro models, mouse models and human patients.
Because uncontrolled cell division is so dangerous for an organism, cells must know not only when to divide, but—crucially—when not to. Cell division arrest prevents tumors and maintains the proper form of tissues. Many cells must also retain the ability to start dividing again when conditions are right, e.g., when the organism must grow, or a damaged tissue must be repaired. A cell in such a temporary, non-dividing state is said to be “quiescent.” Quiescence is a common state for many somatic cells, including fibroblasts, lymphocytes, hematopoietic stem cells, and even dormant tumor cells. Failure to appropriately regulate the transition between quiescence and proliferation underlies several common and lethal disorders, including cancer and chronic wounds. Our research is focused on understanding the molecular basis of quiescence using in vitro models, mouse models and human patients.
Our lab’s research has shown that quiescence isn’t a “sleepy” or default state. In contrast, we found that that quiescence is an active and highly regulated process. We have used sophisticated technologies and computational approaches for understanding the cellular networks that underlie quiescence. We have applied microarrays, mass spectrometry-metabolomics, mass spectrometry-epigenetics and high throughput microRNA profiling to generate and analyze high quality datasets defining the characteristics of proliferating and quiescent cells. We have applied bioinformatic approaches and computational modeling approaches to analyze these datasets to gain greater insight into the biology that underlies quiescence. We have coupled these high-throughput approaches with cell biological, genetic and biochemical strategies to elucidate the functional importance of our findings. In addition, we have established mouse models of injury and tumorigenesis to evaluate our results in the context of normal physiology and pathology.
Through these approaches, in a model system involving primary human diploid fibroblasts, our laboratory’s research has revealed changes in the expression of genes, metabolites, microRNAs, histone modifications, and isoform expression when cells transition between proliferation and quiescence. We discovered that cell cycle arrest with quiescence is not a default state, but rather, is actively maintained by microRNAs and histone modifications that enforce cell cycle exit. We discovered that non-dividing, but not proliferating, fibroblasts actively engage the pentose phosphate pathway to protect themselves from and apoptosis. Our findings also support alternative polyadenylation as a regulatory mechanism that contributes to the transition from a less migratory state in quiescent fibroblasts to a more migratory state in response to proliferative signals.
Areas of Research
.png)
Quiescence
The transition between proliferation and quiescence is frequently associated with changes in gene expression, in the extent of chromatin compaction and in histone modifications, but the role of changes in histone modifications and chromatin state in regulating quiescence is unclear. We are investigating changes in DNA and nuclear structure with quiescence. Specifically, we are investigating the role of the histone methyltransferase Suv4-20h2 in cell cycle regulation.
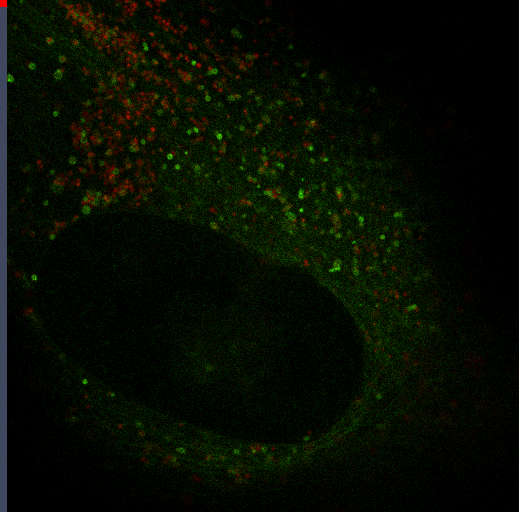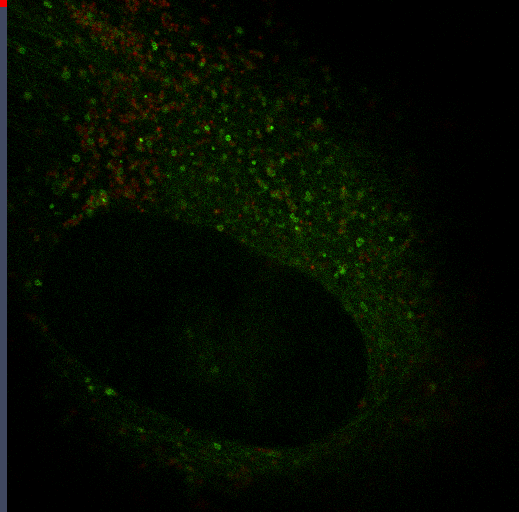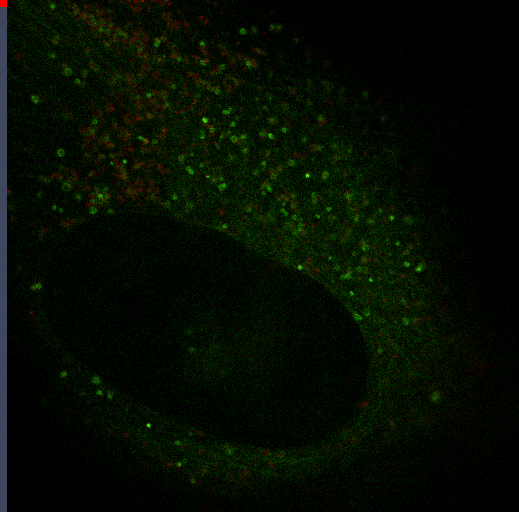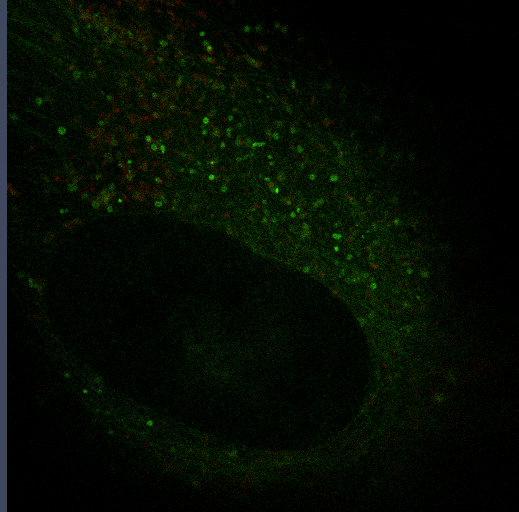
Autophagy
In Wound Healing
Autophagy is an important mechanism for cells to maintain homeostasis through engulfment of proteins and organelles at lysosomes. Using genetically engineered mouse models, immunoblotting, functional depletion, immunohistochemistry and flow cytometry, we are investigating the role of autophagy in the wound healing process.
In Cancer Associated Fibroblast
Autophagy can promote cancer cell survival and growth under stressful conditions. Using genetically engineered mouse models, flow cytometry, functional depletion, immunohistochemistry, and flow cytometry, we have been studying the role of autophagy in the tumor microenvironment.
Cancer Heterogeneity
Tumors from triple negative breast cancer (TNBC) patients are heterogeneous based on genetic variations, tumor histology, and clinical outcomes. We used high throughput genomic data for TNBC patients (n=137) from TCGA to characterize inter-tumor heterogeneity. Similarity network fusion (SNF)-based integrative clustering combining gene expression, miRNA expression, and copy number variation, revealed three distinct patient clusters. Whereas most TNBCs are classified by PAM50 as basal subtype, one of the clusters was enriched in the non-basal PAM50 subtypes, exhibited more aggressive clinical features and had a distinctive signature of oncogenic mutations, miRNAs and expressed genes. Our analyses provide a new classification scheme for TNBC based on multiple omics datasets and provide insight into molecular features that underlie TNBC heterogeneity (Chui et al., Scientific Reports, 2018).
Pancreatic Ductal Adenocarcinoma (PDAC) is an aggressive cancer with a 5-year survival rate of <8%. Unsupervised clustering of 76 PDAC patients based on intron retention (IR) events resulted in two clusters of tumors (IR-1 and IR-2). While gene expression-based clusters are not predictive of patient outcome in this cohort, the clusters we developed based on intron retention were associated with differences in progression-free interval. Network analysis suggested that retention of introns in intron retention cluster 2 could result from disruption of an RBP protein-protein interaction network previously linked to efficient intron removal, that results in included introns and detained transcripts encoding oncogenes in the tumors in this cluster associated with better survival (Tan et al., Genomic Medicine, 2020).